
CRH Resistance: When Your Stress System Stops Responding
By Jacob Gordon, INHC, FMT-CThis article contains affiliate links. As an Amazon Associate, MyBioHack earns from qualifying purchases at no extra cost to you. We only link products we research and stand behind.
Corticotropin releasing hormone (CRH) resistance is a state in which the body's stress signaling system becomes desensitized, dysregulated, or uncoupled, such that CRH stimulation no longer produces the expected hormonal output at the pituitary or downstream tissues, leaving the organism stuck in a pattern of chronic stress with a blunted, exhausted, or paradoxically low response. In this post, we will discuss what CRH is and how the system is supposed to work, what causes resistance, how the receptors and feedback loops break down, which conditions show this pattern, what you can do about it, what to avoid, the mechanisms, and the genetics.
Basics Of CRH And The HPA Axis
Corticotropin releasing hormone (CRH), also called corticotropin releasing factor (CRF), is a 41-amino acid neuropeptide that serves as the master regulator of the stress response in vertebrates. R
It was first discovered and characterized in 1981 by Vale and colleagues, isolated from sheep hypothalamic tissue. R
CRH is synthesized primarily in parvocellular neurosecretory neurons within the paraventricular nucleus (PVN) of the hypothalamus. R
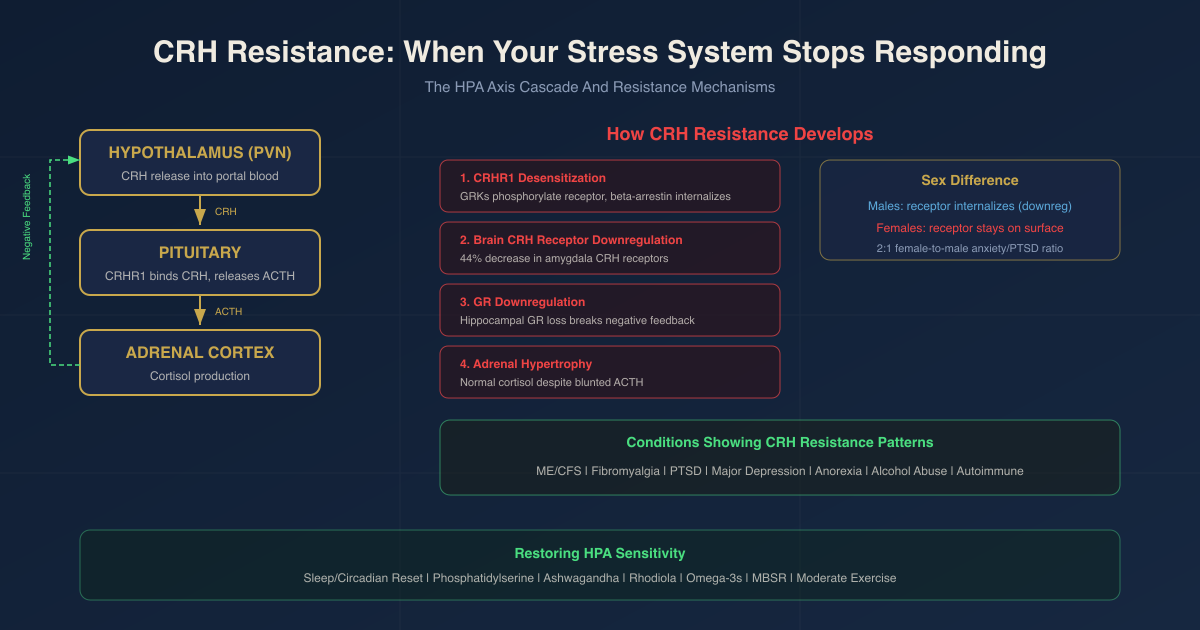
These neurons project to the median eminence, where CRH is released into the hypophyseal portal circulation (a specialized blood supply connecting the hypothalamus directly to the anterior pituitary) to reach corticotroph cells. R
The cascade works like this:
- A stressor (physical or psychological) activates PVN CRH neurons
- CRH is released into the portal blood
- CRH binds to CRHR1 receptors on pituitary corticotrophs
- Corticotrophs release adrenocorticotropic hormone (ACTH) into systemic circulation
- ACTH binds to receptors in the adrenal cortex
- The adrenal cortex produces cortisol (in humans) or corticosterone (in rodents)
- Cortisol feeds back to the hypothalamus, pituitary, and hippocampus to shut down further CRH and ACTH release
This is the hypothalamic-pituitary-adrenal (HPA) axis, and its function depends entirely on the signal fidelity at each step. R
CRH also acts as an extrahypothalamic neuromodulator.
Axons of CRH-expressing neurons project to the amygdala, hippocampus, prefrontal cortex (PFC), bed nucleus of the stria terminalis (BNST), nucleus accumbens, locus coeruleus, and dorsal raphe nucleus. R
In these areas CRH functions as a neurotransmitter modulating fear, anxiety, attention, arousal, autonomic function, and mood, entirely independent of its hormonal role in the HPA axis. R
There are two CRH receptor subtypes:
CRHR1 is the primary stress response receptor. It is highly expressed in the hypothalamus, amygdala, hippocampus, PFC, cerebellum, and pituitary corticotrophs. CRHR1 activation increases HPA axis activity, anxiety-like behavior, and neuronal excitability. Part of this excitability increase comes from CRF directly dispersing gephyrin clusters at inhibitory synapses, reducing the GABAergic and glycinergic brake on postsynaptic neurons. R
CRHR2 has a more restricted and different distribution: the ventromedial hypothalamus, BNST, lateral septum, and peripheral tissues including the heart, lung, skeletal muscle, and gut. CRHR2 mediates stress coping and anxiolysis, opposing or modulating CRHR1 activity. It binds not only CRH but also the related peptides urocortin 2 and urocortin 3 with higher affinity than CRH. R
Normal HPA rhythm:
Cortisol follows a circadian rhythm. It peaks shortly after waking (the cortisol awakening response, or CAR) and is lowest around bedtime. The normal pulsatile rhythm of CRH and ACTH drives this daily cortisol pattern. R
This rhythm is an active, organized output of a well-regulated system. When the system is chronically overburdened, this rhythm is the first thing to go. R
What Causes CRH Resistance
CRH resistance does not usually appear on its own. It develops as a downstream consequence of a system that has been chronically overactivated and has adapted to protect itself from further excess stimulation.
Causes include:
- Chronic psychological stress of any duration (work, relationship, financial, trauma)
- Chronic infections, inflammatory conditions, or autoimmune disease states that persistently activate the HPA axis through cytokine-CRH crosstalk R
- Chronic pain syndromes, which drive persistent CRH and AVP hypersecretion R
- Sleep deprivation and circadian rhythm disruption, which impair the normal CRH pulsatile pattern and negative feedback R
- Childhood adverse experiences or early-life trauma, which alter HPA axis set-points and CRHR1 expression in a durable way through epigenetic mechanisms R
- Prolonged high cortisol from any cause (including exogenous glucocorticoid medications), which downregulates glucocorticoid receptor density at the hypothalamus and pituitary R
- CRH hypersecretion from central CRH-overactivating conditions (melancholic depression, anorexia during active illness, alcoholism during active use), which leads to receptor downregulation R
- Nutritional insufficiency, particularly omega-3 fatty acids, B vitamins, magnesium, and zinc, which impair HPA axis function and feedback sensitivity R
The common thread is that any sustained, unresolved activation of the CRH-HPA cascade eventually causes the system to attempt self-protection through receptor downregulation, feedback uncoupling, and signal desensitization.
How CRH Resistance Develops: The Receptor And Feedback Loop Story
This is the mechanism section that most posts about adrenal fatigue skip entirely, because it is at this level that what is actually happening becomes clear.
Step 1: Receptor desensitization
After CRHR1 or CRHR2 activation, G-protein-coupled receptor kinases (GRKs) rapidly phosphorylate the receptor. This phosphorylation both desensitizes the receptor and increases its affinity for beta-arrestins (adapter proteins). R
Beta-arrestins bind clathrin and beta-adaptin to initiate receptor internalization via clathrin-coated vesicles. The CRHR1 is pulled off the cell surface, reducing signal capacity. R
Internalized receptors can either be recycled back to the surface or degraded in lysosomes. Chronic exposure to high CRH favors the degradation pathway. R
There is a notable sex difference here:
In males, stress increases beta-arrestin2 binding and receptor internalization, leading to CRHR1 downregulation and reduced CRH sensitivity. R
In females, stress prevents CRHR1 internalization, resulting in heightened CRH sensitivity and elevated anxiety-like responses. R
This is one mechanistic reason why women are approximately twice as likely as men to develop anxiety disorders and PTSD.
Step 2: Brain-region-specific CRH receptor downregulation
Chronic high intracerebral CRH levels produce a 44.2% decrease in CRH receptor concentration (Bmax) in the amygdala after just 4 days of chronic CRH pretreatment. R
Critically, pituitary CRH receptor concentration was not significantly altered in the same paradigm. This means the brain-level and pituitary-level regulation of CRHR1 are semi-independent. The amygdala becomes desensitized to CRH (contributing to emotional numbing and reduced stress reactivity) while the pituitary may retain a different response profile. R
This desensitization of the brain CRH processes correlates with the neuroimmune desensitization seen in the same paradigm: the amygdala CRH receptor downregulation temporally correlated with complete abolition of CRH-induced suppression of splenic natural killer cell activity. R
In hippocampus and cortex, CRH downregulates its own receptor (CRF1) via internalization, with a compensatory increase in CRF1 mRNA (the brain upregulates gene transcription to try to replenish surface receptors that are being internalized). R
Pituitary CRF1 expression is modulated by stress in a biphasic manner: a reduction at 2 hours followed by an increase at 4 hours, reflecting the dynamic interplay between desensitization and compensatory upregulation. R
Step 3: Glucocorticoid receptor downregulation and feedback impairment
The HPA axis is supposed to self-regulate through cortisol negative feedback. When cortisol is chronically elevated, it downregulates glucocorticoid receptor (GR) density in the hippocampus and PFC, the key feedback-sensing regions. R
With fewer GRs, these brain regions cannot sense cortisol adequately. They therefore fail to send the "cortisol is high, shut down CRH" signal to the PVN. CRH continues to be secreted at inappropriately high levels despite high cortisol. This breaks the negative feedback loop and perpetuates dysregulation. R
Step 4: Multiple feedback loops entrench the dysregulation
Down-regulation of hippocampal and PVN GRs fails to restrain HPA hyperfunction. R
Persistent HPA activation up-regulates the amygdaloid CRH system, which in turn feeds forward to increase PVN CRH output. R
The hypothalamic and amygdaloid CRH systems form a cooperative, stress-responsive, anxiety-producing neurocircuitry during chronic stress that is responsible for the clinical manifestations of stress-associated disorders. R
Step 5: The glucocorticoid desensitization of CRH neurons themselves
Glucocorticoids rapidly desensitize PVN CRH neurons to norepinephrine (NE) via a receptor-trafficking mechanism. R
Noradrenergic afferents from the locus coeruleus provide a primary excitatory drive to PVN CRH neurons via alpha-1 adrenoreceptors (ARalpha1). During chronic stress, glucocorticoids reroute internalized ARalpha1 from rapid recycling endosomes (which return receptors to the cell surface) to late endosomes and lysosomes (which degrade them). R
This depletes surface ARalpha1 receptors on CRH neurons. CRH neurons become progressively less responsive to NE following repeated acute stress exposures. The net effect: the NE-CRH-ACTH-cortisol axis becomes progressively blunted. R
Step 6: Adrenal adaptation
Long-term CRH hypersecretion in mouse models leads to elevated basal corticosterone, adrenal hypertrophy, and dexamethasone nonsuppression, all reminiscent of the neuroendocrine changes in major depressive disorder. R
The adrenal gland hypertrophies in response to chronic ACTH excess. Paradoxically, this initially allows normal cortisol output despite blunting of the upstream ACTH signal. This is one reason why, in the intermediate stage of HPA dysregulation, you see blunted ACTH with normal cortisol: the adrenal has become more sensitive and efficient because it has been overstimulated for months. R
Conditions That Show CRH Resistance Or Blunted HPA Responses
Chronic Fatigue Syndrome (ME/CFS)
Blunted ACTH responses (P < 0.005) and blunted cortisol responses (P < 0.05) to exogenous CRH were demonstrated in 14 CFS patients free from concurrent psychiatric illness versus 14 healthy volunteers, after administration of 100 mcg ovine CRH. R
The blunted ACTH response to CRH may reflect an abnormality in endogenous CRH levels with a resultant alteration in pituitary CRH receptor sensitivity, or dysregulation of vasopressin or other factors involved in HPA regulation. R
(There is a MAYBE here: other CFS studies have found normal or near-normal ACTH and cortisol responses to CRH challenge, pointing to a heterogeneous patient population and significant methodological variability across studies. The finding is real but not universal.)
Fibromyalgia
40 primary fibromyalgia patients, 28 low back pain patients, and 14 controls underwent a standard 100 mcg CRH challenge test. FM patients displayed a hyperreactive ACTH release in response to CRH challenge (ANOVA interaction effect p = 0.001), alongside mild hypocortisolemia and glucocorticoid feedback resistance. R
This is the opposite pattern from CFS in terms of ACTH (hyperreactive rather than blunted), but shares the hypocortisolemia and feedback resistance, pointing to a dysfunctional cortisol-to-ACTH ratio.
In a separate study, plasma CRH following CRH injection was significantly higher in FM patients and lasted about 45 minutes, paralleled by an increase of somatostatin with a similar time course. Elevated circulating CRH in FM patients suggests elevated levels of CRH-binding protein (CRH-BP), which could explain why ACTH and cortisol do not differ from controls despite elevated CRH, since CRH-BP sequesters CRH and prevents receptor binding. R
PTSD
PTSD patients have increased serum CRH, no clear pattern of change in ACTH, and low cortisol compared to controls. This low cortisol results in increased inflammation, which then acts as a stressor and exacerbates HPA axis dysfunction in a self-reinforcing cycle. R
The likely mechanism: increased GR number and sensitivity in PTSD allows even low cortisol concentrations to adequately suppress CRH and ACTH, producing a paradoxically low cortisol state driven by enhanced negative feedback, not by an exhausted adrenal. R
Major Depressive Disorder
Major depression is characterized by a blunted ACTH response to the CRH test and elevated baseline cortisol. R
CRH-overexpressing mice show elevated basal corticosterone, adrenal hypertrophy, and dexamethasone nonsuppression, changes reminiscent of those reported in MDD, providing a model for how chronic CRH hypersecretion drives the HPA abnormalities seen in depression. R
Blunted ACTH with elevated or normal cortisol is a consistent finding: the adrenal becomes more sensitive to ACTH due to chronic ACTH excess, so less ACTH is needed to drive the same cortisol output. R
Anorexia Nervosa, Alcohol Abuse Disorder, And Pregnancy
All three conditions share a pattern of prolonged HPA over-activation followed by a three-stage recovery: R
Stage 1 (early withdrawal): blunted ACTH + high cortisol.
Stage 2 (intermediate): blunted ACTH + normal cortisol.
Stage 3 (recovery): normal ACTH + normal cortisol.
The mathematical model explaining this: adrenal hypertrophy from chronic ACTH excess allows normal cortisol output even as the upstream ACTH signal attenuates. Recovery of adrenal sensitivity (gland mass normalization) takes weeks to months, which is why ACTH normalization lags behind cortisol normalization. R
Autoimmune And Inflammatory Conditions
Chronic stress leads to glucocorticoid receptor (GR) resistance at immune cells, such that cortisol loses its ability to restrain inflammatory cytokine production (IL-6, TNF-alpha, IL-17) even when cortisol levels are present or elevated. R
This creates a state of simultaneously dysregulated HPA output and impaired cortisol anti-inflammatory action: the cortisol signal is there but the cells have stopped listening to it. R
How To Improve CRH Sensitivity And HPA Function
The goal is not to suppress CRH or cortisol indiscriminately. The goal is to restore the system's ability to respond appropriately, then return to baseline, and then do so again when needed. A dysregulated HPA axis cannot turn off. A recovered one can turn off.
Sleep and circadian rhythm restoration
Dysfunction of the HPA axis at any level (CRH receptor, GR, or mineralocorticoid receptor) disrupts sleep architecture. Conversely, poor sleep perpetuates HPA dysregulation in a bidirectional cycle. R
Restoring consistent sleep-wake timing (same wake time daily, light exposure within 30 minutes of waking) is the single most foundational intervention because it directly resets the PVN CRH neuron circadian clock, which governs when cortisol rises and falls. R
Moderate exercise (not overtraining)
Exercise is a controlled acute stressor that trains the HPA axis to mount a response and then recover. Moderate exercise promotes a healthy cortisol response curve followed by normalization. Overtraining without adequate recovery drives the same chronic dysregulation as psychological chronic stress.
Phosphatidylserine (PS)
Phosphatidylserine is a phospholipid concentrated in the inner leaflet of neuronal cell membranes.
Oral administration of 800 mg/day of brain cortex-derived PS for 10 days significantly blunted both the ACTH (P = 0.003) and cortisol (P = 0.03) responses to physical exercise stress in healthy men, without affecting GH or prolactin. R
Intravenous administration of 50 or 75 mg of brain cortex-derived PS before bicycle ergometer exercise significantly blunted both ACTH and cortisol responses to physical stress. R
600 mg/day of soy-derived PS for 10 days blunted mean peak cortisol concentrations (AUC reduced 35%) and increased the testosterone-to-cortisol ratio, supporting recovery-oriented hormonal balance in male athletes. R
A randomized, placebo-controlled study of 75 healthy men found that 400 mg PS + 400 mg phosphatidic acid (PAS 400) for 3 weeks normalized both ACTH (p = 0.010) and serum cortisol (p = 0.035) responses to the Trier Social Stress Test in chronically stressed subjects. The effect was specific to high chronic stress subjects: low chronic stress subjects showed no significant difference from placebo. R
In a study of chronically stressed men with blunted cortisol response at baseline (the CRH-resistance phenotype), omega-3-enriched PS supplementation appeared to restore (modestly increase) the blunted cortisol response toward normal, rather than suppressing it. This is the bidirectional normalizing effect: PS blunts over-activation and supports under-activation in different baseline states. R
Typical dosing: 400 to 800 mg/day. Best evidence is for 400 to 800 mg/day of soy-derived PS or PS/PA complex for people with active or recent chronic stress.
Ashwagandha (Withania somnifera)
Ashwagandha is the most researched adaptogen for direct HPA axis modulation. Its primary bioactive compounds (withanolides) influence CRH production, GR sensitivity, and adrenal cortisol output through multiple mechanisms. R
A randomized, double-blind, placebo-controlled study of 240 mg/day (Shoden, 35% withanolide glycosides) for 60 days produced a statistically significant reduction in morning cortisol (P < 0.001) and DHEA-S (P = 0.004) in stressed adults, alongside reduced anxiety on the Hamilton Anxiety Rating Scale. R
A 2023 randomized study of 500 mg/day (ARE, standardized to 2.5% withanolides with piperine) for 60 days produced significant reduction in morning salivary cortisol and increase in urinary serotonin compared to placebo, alongside improvements in perceived stress, multitasking, concentration, and decision-making time. R
Systematic review evidence: supplementation with 250 to 500 mg ashwagandha extract daily for 4 to 13 weeks significantly decreased morning cortisol levels in adults with elevated baseline stress. R
The mechanism appears to involve both GABAergic modulation (reducing hypothalamic CRH release) and restoration of glucocorticoid receptor sensitivity at the hypothalamus and pituitary, thereby restoring the negative feedback loop. R
Typical dosing: 300 to 600 mg/day of standardized extract (KSM-66 or Sensoril or equivalent 5% withanolides). Effects are most consistent in high-stress or elevated-cortisol individuals. Less effect in low-stress individuals with normal cortisol.
Rhodiola functions via dual stimulatory-calming effects on the HPA axis, increasing resistance to stressors at sub-maximal activation while normalizing excessive stress responses.
Addressing gut dysbiosis
The gut-brain axis communicates bidirectionally with the HPA axis. Gut dysbiosis can amplify stress responses through inflammatory signaling. Specific probiotics have shown benefits for HPA axis dysfunction by modulating stress responses and cortisol levels, though evidence is insufficient to specify which strains at what doses. R
Addressing dysbiosis through diet and targeted probiotics is a reasonable supportive measure while the primary behavioral and supplement interventions take effect.
Omega-3 fatty acids
Omega-3 fatty acids are critical for HPA axis function and GR membrane sensitivity. Deficiency in omega-3s is associated with impaired HPA axis function and disinhibited cortisol responses. R
Supplementation supports the membrane composition of CRH neurons and corticotrophs, which affects receptor density and signaling efficiency.
Mindfulness-based stress reduction (MBSR)
Meta-analyses confirm that MBSR and other structured mind-body practices produce significant reductions in cortisol, particularly when baseline cortisol is elevated. R
These practices work not only by reducing perceived stress but by reducing the frequency and intensity of CRH neuron firing through top-down PFC regulation of the amygdala-PVN fear circuit.
What To Stay Away From
- Chronic overtraining without recovery drives the same CRH hypersecretion that caused the problem
- Chronic sleep restriction, even by 1 to 2 hours per night, persistently elevates evening cortisol and disrupts the cortisol awakening response R
- High-sugar, high-fat diets exacerbate stress responses and contribute to HPA axis dysregulation by altering CRH and ACTH expression patterns R
- Chronic caffeine abuse, particularly in the afternoon and evening, blunts the cortisol awakening response and disrupts the circadian CRH rhythm
- Prolonged high-dose exogenous glucocorticoids (prednisone, dexamethasone, hydrocortisone), which produce the most reliable and complete GR downregulation at the hypothalamus and pituitary, effectively creating pharmacological CRH resistance
- Alcohol, particularly chronic heavy use, produces the same blunted ACTH, adrenal hypertrophy, and HPA uncoupling seen in anorexia and prolonged stress R
- Chronic psychological isolation, which removes the social buffering of the stress response and keeps the amygdala-PVN circuit chronically activated R
Mechanisms Of Action
Simple:
- CRH is released from the hypothalamus into portal blood in response to stress and binds CRHR1 on pituitary corticotrophs to trigger ACTH and cortisol release R
- Chronic CRH hypersecretion causes GRKs to phosphorylate the CRH receptor, increasing beta-arrestin binding, which pulls receptors off the cell surface via clathrin-mediated endocytosis R
- Fewer surface CRHR1 receptors means each CRH signal produces less ACTH output, creating the blunted ACTH response that defines the post-stress-overload state R
- Simultaneously, excess cortisol downregulates GR density at the hippocampus and PFC, impairing the cortisol feedback brake, so CRH keeps secreting even when cortisol is high R
- Glucocorticoids also reroute norepinephrine receptor trafficking on CRH neurons away from recycling and toward degradation, progressively desensitizing CRH neurons to their primary excitatory input R
- The adrenal gland hypertrophies under chronic ACTH exposure, becoming more sensitive, so cortisol output remains normal (or nearly so) even as the upstream ACTH signal weakens R
- This produces the diagnostic pattern: blunted ACTH after exogenous CRH challenge, with relatively preserved or even normal cortisol output R
Advanced:
Receptor internalization in detail:
CRHR1 is a class B secretin-like G protein-coupled receptor (GPCR). Upon CRH binding it couples primarily with Galpha-s proteins, activating adenylyl cyclase, increasing intracellular cAMP, and activating protein kinase A (PKA). PKA phosphorylates CREB (cAMP response element-binding protein), driving pro-opiomelanocortin (POMC) gene expression and ACTH production. R
In the acute phase, CRH-CRHR1 activation also triggers MAPK/ERK, phospholipase C, and calcium signaling. With repeated activation, GRKs phosphorylate serine and threonine residues in the receptor's intracellular tail. This phosphorylation dramatically increases receptor affinity for beta-arrestin proteins. R
Beta-arrestin acts as a molecular scaffold that recruits clathrin heavy chain and the clathrin adaptor beta-adaptin. These proteins assemble a clathrin-coated pit around the receptor-ligand complex. Dynamin then pinches the pit off the membrane to form an endosome. The receptor is now intracellular. R
From the endosome, CRHR1 can take one of two fates: It can be recycled rapidly back to the plasma membrane via Rab4/Rab11 recycling endosomal pathways, or it can be targeted to late endosomes and lysosomes for degradation. Chronic stress and high sustained CRH favor the degradation pathway, producing net receptor downregulation. R
NR3C1 and GR negative feedback:
NR3C1 is the gene encoding the glucocorticoid receptor (GR). GR is a nuclear receptor. When cortisol binds it, the GR/cortisol complex translocates to the nucleus and binds to glucocorticoid response elements (GREs) in DNA, altering gene transcription. R
In the hypothalamus and hippocampus, GR activation suppresses CRH gene transcription and CRH peptide secretion, completing the negative feedback loop. Chronic cortisol excess downregulates NR3C1 expression in hippocampal neurons, impairing this feedback signal. R
FKBP5 as the GR sensitivity gate:
FKBP5 encodes the protein FKBP51, a co-chaperone of the GR-HSP90 complex. FKBP51 acts as a negative regulator of GR: when FKBP51 binds the GR complex, it reduces GR affinity for cortisol, decreasing receptor sensitivity and impairing translocation to the nucleus. R
Cortisol binding to GR induces FKBP5 gene expression, creating an ultra-short negative feedback loop on GR sensitivity: the more cortisol, the more FKBP51, the less GR responds. In chronic stress, this FKBP51-mediated GR desensitization contributes to glucocorticoid resistance. R
Multiple CRH feedback loops:
Three independent feedback mechanisms cooperate to entrench chronic CRH dysregulation: (1) Attenuation of glucocorticoid-induced negative feedback on hypothalamic and brainstem CRH nuclei activity during chronic stress. (2) Autoregulation of CRH biosynthesis in the PVN via upregulation of CRHR1 within the PVN itself, creating a CRH-to-CRH autocrine loop. (3) Glucocorticoid-mediated pathways that activate rather than suppress amygdaloid CRH, driving an anxiety-amplifying feed-forward circuit. R
Together these loops explain why stress-related HPA dysregulation is self-perpetuating and why single interventions targeting only one node of the system often produce incomplete or short-lived improvement.
Genetics
FKBP5 (rs1360780, rs4713916, rs3800373):
The T allele of rs1360780 in the FKBP5 gene leads to greater FKBP5 induction following GR activation, impairing GR sensitivity and producing prolonged cortisol responses following psychosocial stress. R
T allele carriers with childhood trauma exposure show significant demethylation of the FKBP5 gene, increasing FKBP51 expression, increasing GR resistance, and amplifying the stress response in a way that persists into adulthood through epigenetic memory. R
C allele carriers who experienced similar childhood trauma showed no such demethylation, demonstrating a gene-by-environment interaction where genetic background determines epigenetic vulnerability. R
FKBP5 variants have been associated with increased risk for PTSD (rs1360780, rs4713916), depression, suicidality, and poor antidepressant treatment response. R
In aged individuals (over 50) carrying the T allele, increased cortisol suppression was observed after the Dex/CRH test, suggesting that the functional consequences of FKBP5 variants on HPA reactivity may change with age. R
NR3C1 (glucocorticoid receptor) variants:
NR3C1 encodes the glucocorticoid receptor. Genetic polymorphisms in NR3C1 are a primary regulatory mechanism of GR function and expression. R
Reduced NR3C1 expression (lower GR density) impairs the hypothalamic and pituitary cortisol feedback signal, producing inadequate shutdown of the CRH-ACTH axis and contributing to the sustained CRH hypersecretion pattern. R
Higher GRalpha expression combined with lower FKBP5 expression may serve as a biomarker for chronic depression, reflecting an HPA axis that cannot downregulate adequately. R
Epigenetic modifications including DNA methylation at NR3C1 promoter regions, induced by early-life stress, can lead to lasting changes in GR expression and HPA axis sensitivity that persist across the lifespan. R
CRHR1 variants (rs12938031, rs4792887):
Specific CRHR1 gene variants increase PTSD risk after trauma exposure through impaired cortisol feedback mechanisms. CRHR1 variants interact with childhood adversity to moderate the stress-physical health association. R
Variation in CRHR1 was associated with altered cortisol response to the TSST in a cohort of 368 healthy adults, confirming that CRHR1 genetic variation has measurable effects on HPA reactivity in healthy non-clinical populations. R
The CRHR1-FKBP5 combination is the most studied genetic pair for HPA axis reactivity. Both genes bookend the axis: CRHR1 at the receptor input side, FKBP5 at the feedback output side. Individuals with high-risk variants in both genes have compounded HPA dysregulation vulnerability. R
SLC6A4 (serotonin transporter, 5-HTTLPR):
The short allele of the serotonin transporter gene promoter polymorphism (5-HTTLPR) is associated with HPA axis hyperreactivity in response to social stress. This connects the serotonin system's genetic architecture to HPA axis reactivity, consistent with the known cross-talk between serotonin and the CRH system at the raphe nucleus and BNST. R
More Research
- The sex difference in CRH receptor internalization is underexplored clinically. Females show heightened CRH sensitivity because stress prevents CRHR1 internalization, while males show receptor downregulation because stress promotes it. R This biological divergence may explain the 2:1 female-to-male ratio in anxiety disorders and PTSD, and suggests that sex-specific interventions targeting CRH receptor trafficking rather than just cortisol output could be more effective. This is an active research gap.
- The three-stage HPA recovery model has clinical implications that are not yet widely applied. The mathematical model demonstrating that blunted ACTH with normal cortisol is explained by adrenal hypertrophy rather than CRH resistance at the pituitary level R means that cortisol testing alone at a single time point will miss the HPA uncoupling. A proper CRH challenge test measuring both ACTH and cortisol response curves is needed to characterize the type and stage of dysregulation. This is rarely done in clinical practice.
- CRH-BP as a therapeutic target is unexplored. In fibromyalgia and potentially other hypocortisol conditions, elevated CRH-binding protein sequesters circulating CRH and prevents effective receptor binding, producing the paradox of elevated CRH with blunted downstream ACTH and cortisol. R Interventions that modify CRH-BP levels could normalize the CRH-to-ACTH coupling in these populations without directly manipulating receptor expression.
- Epigenetic reversion of FKBP5 hypomethylation is possible. Early-life trauma demethylates FKBP5, increasing FKBP51 expression and impairing GR sensitivity for decades. R Research on whether sustained psychotherapy, EMDR, or other interventions can reverse this methylation change is ongoing, and early evidence in PTSD treatment is promising. Understanding the epigenetic dimension of HPA resistance may eventually explain why some people respond well to behavioral interventions while others require pharmacological targeting of the GR-FKBP51 pathway.
Jacob Gordon
INHC, FMT-C
Board Certified Health Coach
I spent years battling unexplained chronic illness before discovering biohacking, epigenetics, and functional medicine. Now I share that research at MyBioHack to help others find their own answers.
Book a ConsultationRelated Protocols & Supplements
Deep-dive chapters and recommended supplements for this topic
Lion's Mane
1000mg/day
Omega-3 (DHA)
2g/day
Phosphatidylserine
100mg 3x/day





